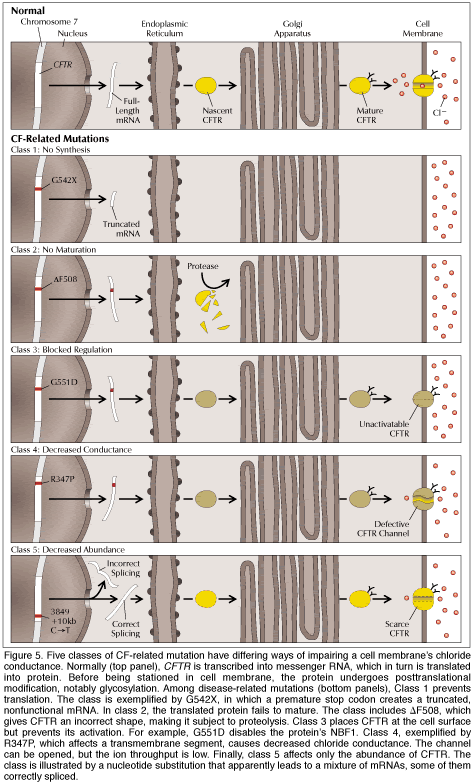
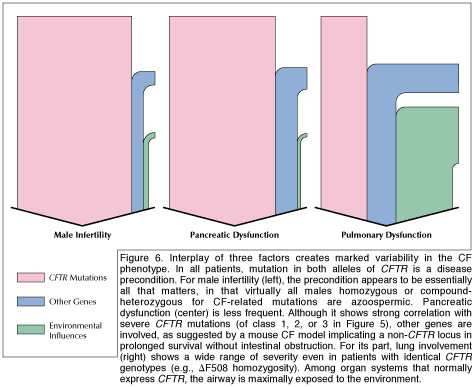
|
Caption reads: Gentle pounding on the chest, or chest percussion, has long been a standard treatment for cystic fibrosis. This procedure aims to clear mucus from clogged airways in the lungs. Investigators hope that growing understanding of the molecular basis of the disease will lead to drug therapies that prevent airway obstruction in the first place. The child here is being tapped by her mother. The white unit on her arm delivers intravenous antibiotics to combat infection of the lung. |
| http://www.wellesley.edu/Chemistry/chem227/lipids/cf-transport.htm |
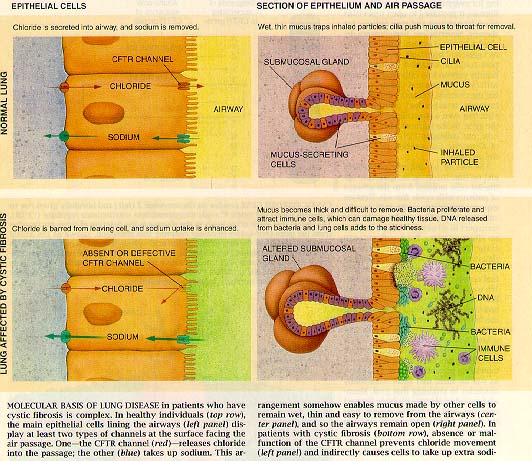 |
Caption reads: Molecular basis of lung disease in patients who have cystic fibrosis is complex. In healthy individuals, the main epithelial cells lining the airways display at least two types of channels at the surface facing the air passage. One - the CFTR channel - releases chloride into the passage; the other (blue) takes up sodium. This arrangement somehow enables mucus produced by other cells to remain wet, thin and easy to remove from the airways and so the airways remain open. In patients with cystic fibrosis, absence or malfunction of the CFTR channel prevents chloride movement and indirectly causes cells to take up extra sodium. |
| http://www.wellesley.edu/Chemistry/chem227/lipids/cf-transport.htm |
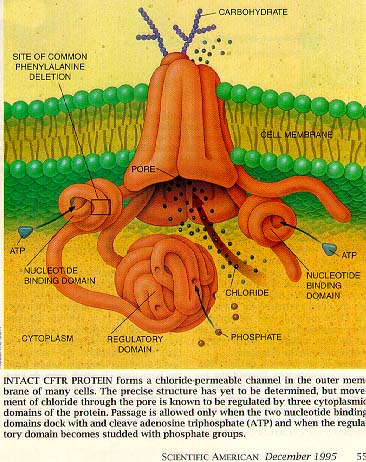
|
Caption reads: Intact CFTR protein forms a chloride-permeable channel in the outer membrane of many cells. The precise structure has yet to be determined, but movement of chloride through the pore is known to be regulated by three cytoplasmic domains of the protein. Passage is allowed only when the two nucleotide-binding domains dock and cleave adenosine triphosphate (ATP) and when the regulatory domain becomes studded with phosphate groups. |
| http://www.wellesley.edu/Chemistry/chem227/lipids/cf-transport.htm |
 |
Cystic fibrosis affects the lungs, pancreas, gastrointestinal tract, salivary and sweat glands and reproductive organs. The CF gene was discovered in 1989 by Lap-Chee Tsui, John R Riordan, and Francis S Collins. CF is an autosomal recessive disease. Traits skip a generation and equal number of males and females are affected. If both parents are affected, all offspring will be affected. Most affected people have unaffected parents. Tests include testing for elevated levels of trypsinogen, which is characteristic. There is no cure. |
| www2.carthage.edu/~pfaffle/ hgp/Tech.html |
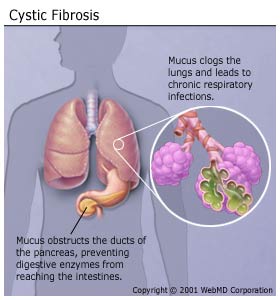
|
People who have CF produce abnormally thick, sticky mucus, which clogs the lungs and leads to recurring lung and sinus infections and difficulty breathing. Reduced oxygen in the blood also leads to a characteristic rounding and enlargement of the nail bed in the fingers and toes, called clubbing. Those with the disease may also develop a barrel-shaped chest as a result of their increased work to breathe. These repeated infections often lead to fleshy growths inside the nose, called nasal polyps. The thick mucus also obstructs the ducts of the pancreas, preventing digestive enzymes from reaching the intestines. So those with CF do not absorb nutrients from their food well, and they eliminate nondigested food through the bowel, resulting in very large stools. Because so little food is absorbed, those with CF have difficulty gaining and maintaining weight, despite a healthy appetite and diet. CF also affects the reproductive systems of both males and females. Although females with CF have normal fallopian tubes and ovaries, their thick cervical secretions may block sperm entry and prevent them from getting pregnant. Males with CF are almost always sterile because they produce relatively few or no sperm. Also, abnormally thick secretions may block the ducts that carry sperm, or the ducts may not develop properly. Another hallmark of CF is an unusually high concentration of sodium and chloride (salt) in the sweat. The defective CF gene causes faulty chloride movement in CF cells. As a result, parents often report that their babies "taste very salty" when they kiss them. |
| http://www.hunterdonhealthcare.org/webmd/topics/cystic_fibrosis/basics.asp |
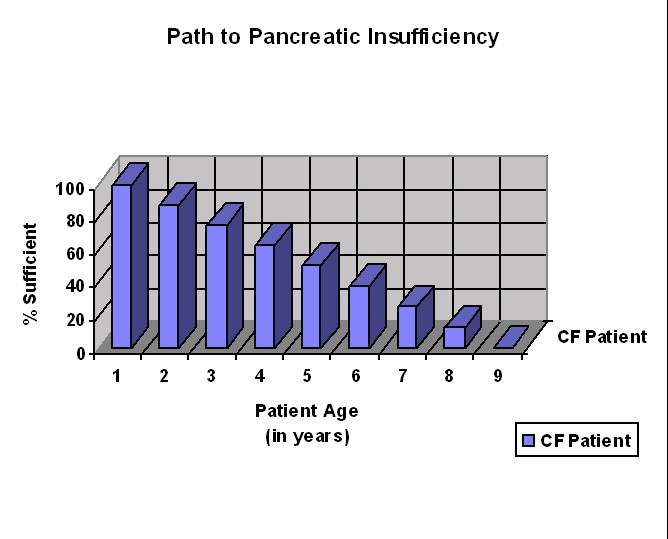
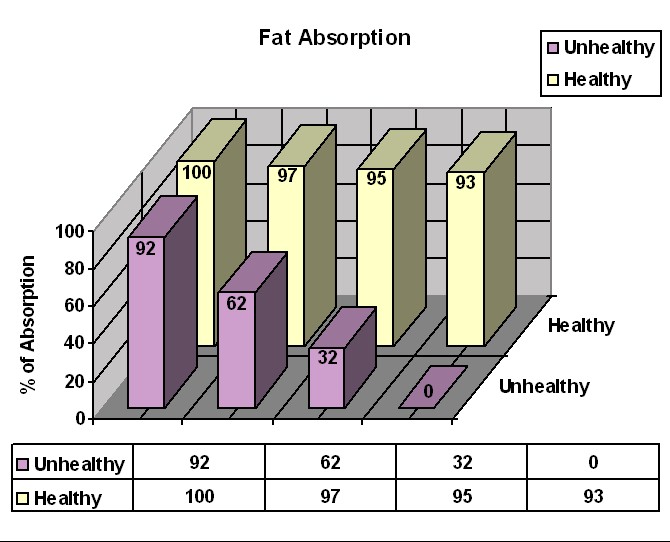
Before effective treatment was available, most CF children died in infancy, in part from starvation (malnutrition) due to their pancreatic insufficiency. The human body has a number of redundant systems that allow itself to continue functioning when a certain system in the body does not function properly. When the pancreas does not produce enough enzymes to digest the food in the small intestine, the intestinal cells have a backup group of enzymes that do fairly well at digesting starch and protein. In this case, however, the digestion and absorption of starch is not complete, and the remaining starch may cause gas when it gets into the large intestine. The digestion of protein is also not complete, so that some patients, especially infants, may have low levels of protein in the blood. If protein levels are sufficiently low, fluid may leak out of the blood vessels and cause puffy skin (edema).
|
|
|
While the intestinal cells can deal with, in part, starches and proteins, these cells do not have a backup means to digest fat, and the effectiveness of the lipase produced by the salivary glands is limited. Therefore, most CF patients do not digest and absorb fat well, and fat is passed out of the body in the bowel movements. Fat makes the bowel movements large, greasy, and more smelly than normal, which is one of CF's characteristic traits. Furthermore, all of the fat that comes out in the bowel movements is lost to the body. A given amount of fat has more calories than any other kind of food, so losing an ounce of fat means losing more than twice as many calories as would be lost in an ounce of carbohydrate or protein. Loss of fat in the bowel movements thus leads to malnutrition and poor growth, despite a huge appetite. The "textbook picture" of a child with undiagnosed CF is someone who is scrawny, has a huge appetite, and has frequent, large, smelly, greasy stools. This person is scrawny because most of the nutrients in the food consumed go directly into the toilet. The large appetite is really a way of compensating for losing half of what is eaten in the bowel movements - it's as if you've been fed a half portion, so you eat another portion to make up for that. Lack of fat absorption may also lead to the lack of special kinds of fatty nutrients that are essential to health - "essential fatty acids", and fat-soluble vitamins (vitamins A, D, E, and K).
|